|
Fifth Dimension Catalog Contact Us |
||||||||

|
Impact of aging on the biology of breast cancer |
|||||||
| ||||||||
4. Cellular mechanisms linking aging and cancer
Back to the Table of Contents
Geroscientists contend that among the causes of aging in mammals (and virtually all multicellular organisms) are evolutionarily conserved cellular responses designed to protect an organism from developing cancer. These genome-protecting and tumor suppressing mechanisms include apoptosis and senescence, cellular responses that effectively eliminate or prevent proliferation of genomically damaged but otherwise replication-competent somatic cells at risk for neoplastic transformation. [16] The tumor suppressing proteins p53 and p16INK4a (product of the INK4a/ARF locus) have been linked to both aging and tumorigenesis in animal models;[17,18] and it has recently been argued that these two key regulators direct convergent and divergent cellular mechanisms, respectively, that evolved to protect against cancer and aging.[19] Convergent mechanisms, including improved metabolic efficiency, antioxidant defenses and p53 transcriptional responses, act to diminish cellular damage and simultaneously protect against cancer and aging; divergent mechanisms, including telomere shortening and derepression of INK4a/ARF (producing p16INK4a overexpression), act mainly to reduce cellular proliferation and prevent tumorigenesis but in so doing promote aging by limiting the regenerative potential of stem cell populations.[19] Studies of human aging syndromes (progerias) suggest that various other genes also regulate both aging and cancer. [20] None of the well-studied progeroid syndromes (Hutchinson-Gilford, Werner, Bloom, Rothmund-Thomson, Cockayne, dyskeratosis congenita, trichothiodystrophy) are thought to perfectly represent precocious total body aging; in fact, many involve only segments of body aging and are thus referred to as segmental progerias. Genotoxic stress in the form of unrepaired DNA damage, caused by physical (e.g. UV, X-rays) or chemical (e.g. reactive oxygen species) agents, some from our own cellular metabolism, can produce DNA mutations and chromosome aberrations that lead to cancer and/or trigger cell senescence or apoptotic mechanisms that promote aging by causing functional decline and loss of organ or tissue cellularity. The rate and type of DNA damage (e.g. single strand or double strand breaks, base adducts or interstrand crosslinks) as well as a cell's ability to respond and repair this damage determine the cellular and organismal consequences: cancer, aging or both. An intricate network of repair systems have evolved to address specific subclasses of DNA damage (e.g. nucleotide and base excision repair, transcription-coupled repair, homologous recombination and non-homologous end-joining to fix double strand breaks), producing a fine balance between anti-cancer and anti-aging protection mechanisms.[21] When unrepaired in proliferating cells, DNA damage may be either diluted out by replication or propagated into mutations and chromosome aberrations within daughter cells, resulting in malignant transformation; when unrepaired in non-dividing (postmitotic) cells, DNA damage may gradually accumulate until cell death or senescence ensues. Varying disturbances in the balance between anti-aging and anticancer genome maintenance mechanisms are apparent in the different progeroid syndromes. Those syndromes associated with increased risk of malignancy (e.g. Werner, Bloom, Rothmund-Thomson, xeroderma pigmentosa, dyskeratosis congenita) often result from inherited mutations in genes involved in global repair systems (e.g. DNA helicases), generating increased genomic mutagenesis. In xeroderma pigmentosa, for example, the resulting increase in mutagenesis produces a 1000-fold propensity for skin cancer formation yet only minor symptoms of premature aging. In contrast, syndromes with genetic defects in more localized DNA repair systems that do not prevent mutations but promote cell death or senescence responses (e.g. Cockayne syndrome and trichothiodystrophy) exhibit many symptoms of premature aging but may be associated with a decreased likelihood of cancer.[21] Curiously, the ratio between cancers of epithelial origin and sarcomas of mesenchymal organ in the general population is about 10:1, yet in conditions like Werner syndrome this ratio is 1:1 suggesting that mesenchymal tissues are more susceptible than epithelial tissues to the consequences of an inherited deficiency in genome maintenance. It is also puzzling that the genes mutated in Werner (WRN) and other progeroid syndromes have not been observed to be mutated or lost in either inherited or sporadic forms of breast cancer.[20]
4.1. Do late-onset breast cancers derive from senescent
stroma or epithelium?
Cellular senescence was first described over 40 years ago as a process limiting the proliferation of normal human cells. Today, this specific phenomenon is termed replicative senescence and is thought to be triggered by progressive telomere shortening. However, other stressful stimuli (e.g. DNA damage from ionizing radiation or drugs, inappropriate mitogenic signaling, oxidative stress) can also readily induce the senescent cell phenotype, triggered by p16INK4a and/or p53 activation. The senescent cell phenotype is generally described as irreversible proliferation arrest with resistance to apoptosis and altered cell function, including increased secretion of degradative enzymes, inflammatory cytokines and growth factors.[16] It has been proposed that senescent cells slowly accumulate with age; indeed, cell senescence is thought to contribute to aging while protecting from tumorigenesis.[16,22]
Thus, to fully transform a population of senescent epithelial cells, the tumor suppressing functions of p16INK4a and p53 must be bypassed. Certainly
telomere attrition is observed as an early event in breast tumorigenesis, correlating with increased genomic instability;
however, there is considerable variation in telomere length among fully formed breast cancers, and the silencing
of p16INK4a and/or mutation of p53 are seen in only a proportion of all breast cancer cases.[23]
Primate studies have shown that senescent fibroblasts accumulate with advancing age, however, in postmitotic tissues there is little evidence of age-related senescence.[24] The extent to which senescent fibroblasts or epithelial cells accumulate within an aging breast is presently unknown; but there is ample experimental evidence to suggest that an aging stoma can promote breast tumorigenesis, largely by remodeling the extracellular matrix and promoting invasion and growth of premalignant epithelial cells exposed to the secretory products of the senescent fibroblasts.[16,25,26]
Thus, it seems ironic that while the senescence response appears designed to protect a cell population from malignant transformation,
senescent stroma can promote tumorigenesis of neighboring premalignant or malignant epithelium. Further
insights are needed into the multi-faceted cancer-senescence relationship, as more recent experimental evidence suggests
that tumors formed in the absence of functioning p53 can be ablated by reactivation of p53, which induces
tumor cell senescence sufficient to arrest tumor growth followed by macrophage and immune cell induced tumor cell destruction.[27]
4.2. Do cancer-aging hypotheses predict clinical breast cancer behavior?
Observations of age-dependent deterioration in genome integrity along with increased gene silencing by promoter
methylation continue to fuel speculation that genetic and epigenetic aging events drive the increasing cancer incidence of later life.[11]
Normal human aging appears to be associated with telomere shortening and increased genomic instability, global and promoter-specific epigenetic changes, and altered
expression of genes involved in cell division and extracellular matrix remodeling [28-31] -characteristics shared by
many epithelial malignancies like breast cancer. Thus, cancers increased with aging are thought to possess a mutator
phenotype predisposing to genetic instability, accelerated proliferation, and a generally more invasive and metastatic
phenotype.[11] From a clinical perspective, however, there
is little direct evidence to support this mechanistic paradigm; and for breast cancers in particular, there is definitive evidence
to the contrary. Clinical observations in older patients indicate that their tumors grow more slowly and are biologically
less aggressive.[32,33] Also, early-onset breast
cancer is known to be clinically more aggressive than late-onset breast cancer;[5] and younger age (<45 years) has
been shown to be an independent risk factor for early breast cancer recurrence and death.[34,35] To confirm such observations
within a histologically identical group of early-stage ER-positive breast cancers, we turned to a colleague (A. Thor, MD) possessing a well studied archive of >800 breast cancers associated with 18+ years of clinical follow-up
and fully characterized by various prognostic markers. [36]
Selecting for untreated ER-positive node-negative (T1/2) ductal carcinomas diagnosed before age 46 or after age 69
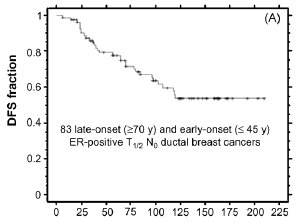
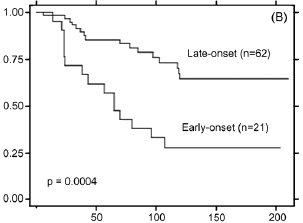
yielded only 83 eligible cases (21 early-onset, 62 later-onset). However, as shown by the Kaplan-Meier plots in Fig. 2, long-term disease free survival (DFS) was significantly different between the two age groups, with 10-year DFS plateauing
at <30% of the early-onset group and >70% of the later-onset group (p = 0.0004). Adjusting for differences in tumor
grade and proliferative index (Ki67/MIB-1) between the two ER-positive tumor groups failed to eliminate the significant
outcome differences, supporting the contention that unknown biological features determine the different clinical behaviors
of histologically similar early-onset and late-onset breast cancers.
5. Biological differences between early-onset and
late-onset breast cancers
Back to the Table of Contents
To test the premise that breast cancer biology is age-dependent, we performed a retrospective analysis on nearly 4000 primary breast cancers, derived from two geographically different archives (American/MGH, Swiss) and previously characterized with respect to multiple validated prognostic and predictive biomarkers.[36] The paraffin archived American samples were analyzed for histology, tumor grade, stage (TNM), apoptotic and mitotic indices, and were immunohistochemically scored for Ki67/MIB-1, p53, ERBB2, EGFR, ER, PR, and pS2. In addition to scoring tumors by histology, grade and stage, protein extracts from the cryobanked Swiss samples were quantitatively analyzed by immunoassays for ERBB2, EGFR, ER, PR, pS2, Bcl2, VEGF, uPA, uPAR, PAI-1, and cathepsin D. In aggregate, these biomarkers represent surrogate measures of tumor (i) growth, proliferation, and genetic instability, (ii) angiogenic, invasive and proteolytic potential, and (iii) endocrine dependence. Findings from both archives demonstrated that late-onset breast cancers have slower growth rates, are genomically more stable and more likely to be ER-positive, and are less likely to be ERBB2-positive or EGFR-positive. Altogether, they support the conclusion that the biology of breast cancer is age-dependent; however, they do not account for the strong inverse interactions observed between ER and the other age-dependent biomarkers.
Months |
Months |
Fig. 2. Kaplan-Meier disease-free survival (DFS) curve for combined set of 83 ER-positive node-negative ductal breast cancer cases untreated with adjuvant therapy (panel A), and DFS curves for late-onset (n = 62) and early-onset (n = 21) subsets (panel B). As described in the text, the selected age cohorts were well matched for numerous tumor characteristics and biomarkers and differed only by mean tumor proliferation index and high tumor grade. The significant difference in DFS outcomes shown (p = 0.0004) could not be eliminated by adjusting for subset differences in tumor grade and proliferation index. Primary data provided by A. Thor and analyzed by D. Moore.
5.1. Inverse age relationship between ER and measures
of breast cancer growth and genome stability
All surrogate measures of tumor growth and genetic instability showed strong inverse correlations with ER and patient
age at diagnosis, when evaluated on a decade-by-decade
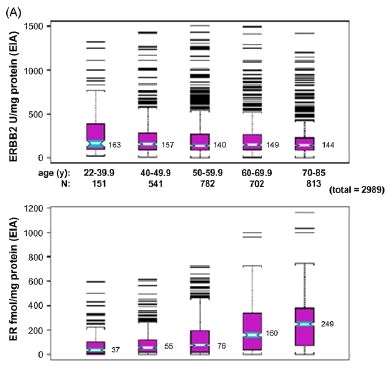
basis.[36] As shown in Fig. 3, across both archives and
whether evaluated quantitatively (panel A) or immunohistochemically (panel B), overexpression of the ERBB2 growth
factor receptor declined significantly after age 40, while total ER content and the proportion of ER-positive breast cancers
increased continuously after age 40. A similar relationship was seen for the EGFR growth factor receptor. Relative to
aging normal mammary gland tissue, these age-dependent changes in breast cancer ER content (fmol/mg protein) mirrored
10-fold lower increases in normal mammary gland ER content up to age 60, rising faster thereafter and reaching
a near 25-fold differential between malignant and normal
breast tissue by age 80.[4],[36] Also showing inverse relationships
to ER content, breast cancer p53-positivity and apoptotic index declined fastest after age 50, while grade,
mitotic index and Ki-67/MIB-1 declined most rapidly prior to age 60.[36] These age-dependent biomarker changes seen
in nearly 4000 unselected breast cancer cases were therefore consistent with both clinical and epidemiological evidence
indicating that early-onset breast cancers are more aggressive than late-onset breast cancer cases.[5],[34,35] Furthermore,
they clearly demonstrated the strong inverse age relationships between breast cancer ER content and all surrogate measures
of breast cancer growth and genetic instability.
5.2. Aging and measures of breast cancer invasiveness and angiogenesis
Analysis of both breast cancer archives indicated that after age 40 there was no consistent age relationship with tumor
stage (TNM staging), nodal involvement, or risk of distant metastasis (M1 stage) at the time of diagnosis.[36] Validated
prognostic and predictive biomarkers associated with subsequent risk for local, regional or systemic dissemination
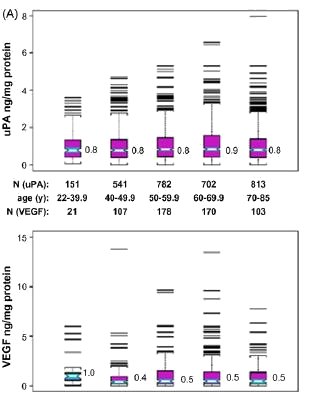
include the angiogenic growth factor, VEGF, and the secreted proteases, uPA and cathepsin D. As illustrated in Fig. 4 (panel
A), none of these surrogate measures of invasive or metastatic potential showed any significant change when analyzed on
a decade-by-decade basis in breast cancer cases diagnosed after age 40, although tumor VEGF levels were on average
two-fold higher in tumors arising before age 40 than in those arising after age 40.[36] While expression levels of these
biomarkers mediating breast cancer invasiveness and angiogenic potential did not change significantly with increasing
age, a more recent study suggested that similar tumor expression of VEGF or uPA might be associated with significantly
different clinical outcomes (relapse-free survival) when comparing late-onset and early-onset breast cancers of similar type.[37] As shown in Fig. 4 (panel B), when transcript levels
of VEGF and uPA were assessed in a different population of late-onset (>= 70 years, n = 25) and early-onset (<= 45 years,
n = 29) node-negative ER-positive breast cancer cases, higher levels of either VEGF or uPA expression were associated with
significantly more relapses in the early-onset cases but were not as prognostic in the late-onset cases, despite comparable
expression levels of VEGF and uPA in both age cohorts. It will be important to discern if such age-specific outcome differences,
in the absence of intrinsic differences in prognostic biomarker tumor expression levels, can be confirmed in future
studies. If so, such observations would point to important age-specific differences in clinical susceptibility to biologically
similar breast tumors.
 |
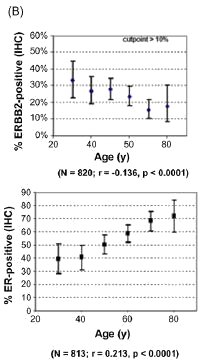 |
Fig. 3. Age associations for ERBB2 and ER content (panel A) or their percent overexpression (panel B) for unselected primary breast cancers from two different archives. Cryobanked Swiss tumor extracts (n = 2989) were analyzed by quantitative enzyme immunoassays (EIA), while formalin-fixed paraffin-embedded American/MGH samples (n > 800) were analyzed by immunohistochemistry and scored for percent positive staining tumor cells. Notch-boxplots show median values for each age group. Proportion plots show median% values (±95% confidence intervals) for each age group, with linear regression fit (r, Pearson's correlation coefficient) and statistical significance (p values) indicated below. Figures modified from previous publication. [36]
 |
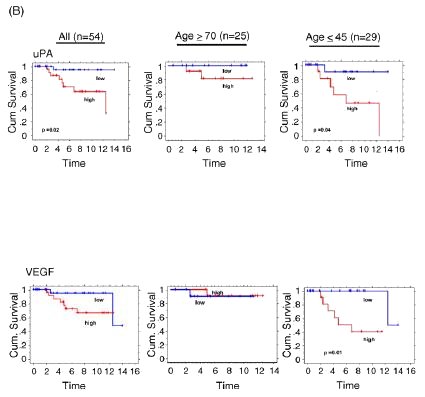 |
Fig. 4. Age associations for uPA and VEGF protein content from Swiss breast cancer archive (panel A), and Kaplan-Meier relapse-free survival curves based on level of breast cancer uPA and VEGF transcript expression (high, low) from American/UCSF breast cancer archive (panel B). Unselected Swiss samples were assayed as described in Fig. 3 (panel A), with figures modified from previous publication.[36] American/UCSF archive contained 54 node-negative, ER-positive breast cancer cases selected according to late-onset (>= 70 years, n = 25) or early-onset (<= 45 years, n = 29). Dichotomization for uPA and VEGF expression levels (high, low) was based on mean-centered transcript values, measured as previously described; figures were modified from previous publication.[37] Significant differences between the cumulative survival curves were determined by Log Rank analyses (only p values <0.05 shown).
5.3. Early-onset and late-onset breast cancers arise by
epigenetically different mechanisms
The strong inverse age relationships observed between breast cancer ER content or positivity and the multiple indices
reflecting breast cancer growth and genetic instability raised concerns about the relative importance of age versus ER status
in determining breast cancer biology. Several prospective studies were recently initiated to address this issue, using
early-onset (<= 45 years) and late-onset (<= 70 years) breast cancer specimens of known ER status, derived from two
independent breast tumor cryobanks (American/UCSF, Italian).
DNA extracted from these samples was analyzed for p53 mutations (exons 5-8) and whole genome aberrations by array comparative genomic hybridization (CGH), while
RNA extracted from these samples was analyzed by high-throughput expression microarrays, performed as previously
described. [23], [38] Table 1 shows the frequency of wild-
type p53 (p53wt) versus mutated p53 (p53mut) found in ER-positive and ER-negative subsets of early-onset (n = 135)
and late-onset (n = 154) breast cancers, regardless of tumor stage. The most significant differences to be noted are
that late-onset breast cancers are 1.5-fold more likely to be ER-positive/p53wt and 0.45-fold as likely to be ER-negative/p53wt as compared to early-onset breast cancers.
While p53 mutations are much less frequently found in ER-positive as compared to ER-negative breast cancers, it is
surprising to discover that when ER status is controlled for, p53 mutations are not significantly more frequent in early-
onset breast cancers relative to late-onset breast cancers.[39]
Likewise, when array CGH changes were compared between 27 early-onset and 44 late-onset ER-positive ductal breast
cancer cases, the two most commonly observed ER-positive breast cancer genotypes (1q gain/16q loss and amplifier genotypes)
were equally represented in both age cohorts;[23],[40]
and no significant age differences were apparent in any of the observed genome-wide aberrations, including frequencies
of the most common breast cancer amplicons (e.g. ERBB2 amplicon: 11% in early-onset, 5% in late-onset). In
contrast, when expression microarray changes were compared between 53 early-onset and 48 late-onset ER-positive
node-negative breast cancer cases, both unsupervised and supervised analyses of the 5.1K variably expressed genes
identified significant age-specific differences.[40] Unsupervised
analysis revealed that ER-positive breast cancers are heterogeneous and comprise as many as six different
transcriptome subtypes including two with a significant age bias. Supervised analyses revealed that late-onset ER-
positive breast cancers express significantly higher levels of ER transcripts as compared to early-onset ER-positive cases;
increased levels of some tumor suppressors, developmental regulators, and apoptosis inducers; and decreased levels of
specific growth regulators and mitotic factors. These findings provide a new mechanistic basis for claiming that when
ER status is controlled for, early-onset breast cancers exhibit much greater proliferative potential than late-onset breast
cancers, potentially explaining in part their earlier clinical appearance.[40] While it is surprising that early-onset breast
cancers appear to lack significant genomic differences from late-onset breast cancers, there appear to be sufficient epigenetic/transcriptome differences to conclude that when ER
status is controlled for, late-onset and early-onset breast cancers arise by fundamentally different biological mechanisms.[40] Other age cohort studies of this design and type are
now needed to further generalize about potential age-related biological differences driving ER-negative breast tumorigenesis,
as well as the many other age-associated epithelial malignancies besides breast cancer.
| Table 1 | ||||
| Frequency of wildtype p53 (p53wt) versus mutated p53 (p53mut) found in ER-positive (ERpos) and ER-negative (ERneg) subsets of early-onset and late-onset breast cancers | ||||
| n = 289 | ERneg/p53wt (%) | ERpos/p53wt (%) | ERneg/p53mut (%) | ERpos/p53mut (%) |
| Late-onset (>= 70 years), n = 154 | 25 (16.2%) | 107 (69.5%) | 12 (7.8%) | 10 (6.5%) |
| Early-onset (<= 45 years), n = 135 | 49(36.3%) | 64 (47.4%) | 14 (10.4%) | 8 (5.9%) |
| p = 0.0004, Fisher Exact. | ||||
6. Conclusions
Back to the Table of Contents
Whether the molecular and cellular effects of normal mammary gland aging produce background effects from which breast malignancy must be differentiated or in some way contribute to the breast carcinogenic process remains a question of fundamental importance. Clinical observations and biomarker studies indicate that late-onset breast cancers grow more slowly and are biologically less aggressive than early-onset breast cancers, even when controlled for ER receptor expression, supporting the conclusion that the biology of breast cancer is age-dependent. Initial studies comparing early-onset and late-onset ER-positive breast cancers for DNA mutations and whole genome aberrations as well as RNA transcriptome differences suggest that epigenetic changes rather than genotypic variation account for most of the age-dependent biological and clinical differences observed in hormone-dependent breast cancer.
Please cite this article in press as: Benz CC, Impact of aging on the biology of breast cancer, Crit. Rev. Oncol./Hematol. (2007), doi:10.1016/j.critrevonc.2007.09.001
|
You are welcome to share this © article with friends, but do not forget to include the author name and web address. Permission needed to use articles on commercial and non commercial websites. Thank you. |